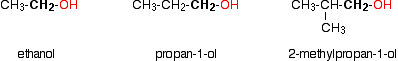
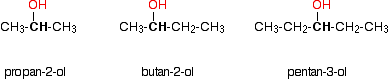
Monohydric alcohols can be classified into three types as primary, secondary and tertiary.
Primary alcohols

Secondary alcohols
Tertiary alcohols
Physical properties
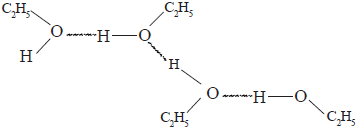
In alcohols the –OH bond is polarized as – R – O δ- -H δ+ . Hence, due to the inter molecular hydrogen bonds formed between alcohol molecules, their boiling points have higher values compared to the alkanes and ether with comparable relative molecular masses. The boiling point increases in going down the series of alcohols.
![]()
The above diagram shows how the inter- molecular hydrogen bonds exist in ethanol. Alcohols which have low relative molecular mass are soluble in water. The solubility of alcohols in water is due to the – OH group which can forms H – bonds with water molecules. The non polar alkyl group in the alcohol molecule is a hindrance to the solubility in water. In going down the homologous series of alcohols the size of the non- polar alkyl group gradually increases relative to the –OH group.
Reactions involving cleavage of the O-H bond
(i) Reaction with sodium
Alcohols behave as acids and react with sodium liberating hydrogen and forming sodium alkoxides. The alkoxide ion is a strong nucleophile and also a strong base.
(ii) Reaction with carboxylic acids (Acylation of alcohols)
Alcohols react with carboxylic acids to form esters. For this esterification reaction, concentrated H2SO4 acid acts as a catalyst.
The esterification reaction is both slow and reversible. The equation for the reaction between an acid RCOOH and an alcohol R’OH (where R and R’ can be the same or different) is:


So, for example, if you were making ethyl ethanoate from ethanoic acid and ethanol, the equation would be:


Nucleophilic substitution reactions that take place by the cleavage of C-O bond
(i)Reaction with PCl3 or PCl5
Alcohols react with PCl3 or PCl5 to give alkyl chlorides.


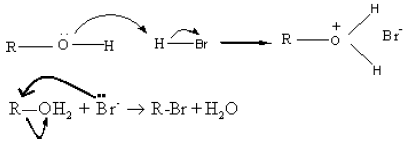
(ii) Reaction with hydrogen halides
![]()
Alcohols under go nucleophilic substitution reaction with HBr to give the corresponding alkyl bromides. Protonation of the O atom converts the -OH group into a better leaving group.
![]() In this reaction Br- ion acts as a nucleophile and the leaving group is H2O.
In this reaction Br- ion acts as a nucleophile and the leaving group is H2O.
(iii) Reaction with anhydrous ZnCl2 and conc. HCl (Lucas test)
In this reaction, R-OH is converted to R-Cl. ZnCl2 is used as a catalyst. Because alkyl halides are insoluble in water, as the raction proceeds the reaction mixture becomes cloudy and turbid. The time taken for the turbidity to appear, after the mixing of reagents, can be used to distinguish between primary, secondary and tertiary alcohols.
Under the provided reaction conditions the above nucleophilic substitution reaction takes place in two steps. Tertiary alcohols form stable intermediate tertiary carbocations and therefore, tertiary alcohols in the presence of the Lucas reagent forms a turbidity in a very short time. Secondary alcohols take longer time and primary alcohols react very slowly.
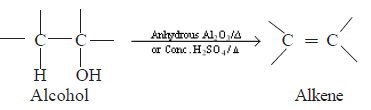
Elimination reactions
Alcohols undergo an elimination reaction when treated with conc. H2SO4 or when heated with alumina to a higher temperature. The reaction in which a molecule of water is eliminated from an alcohol is the dehydration of alcohols. Here, an alkene is formed as the product of the reaction.
![]()
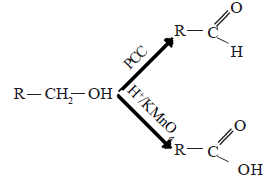
Oxidation of alcohols
The product of oxidation depends on primary, secondary or tertiary nature of the alcohol. Oxidation of alcohols can be carried out with H+/KMnO4 or H+/K2Cr2O2 or H+/CrO3.
(i) Primary alcohols

In the presence of the above mentioned oxidizing agents primary alcohols first give aldehydes. These are further oxidized to carboxylic acids. If pyridinium chlorochromate [C5H5NH+CrO3Cl] is used, the reaction can be stopped at the stage where aldehyde is formed.
![]()
(ii) Secondary alcohols are oxidized to give ketones.
![]()
(iii)Tertiary alcohols
Normally the tertiary alcohols do not undergo oxidation under conditions that primary and secondary alcohols are oxidized.
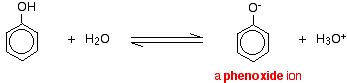
Aromatic compounds, in which an -OH group is joined directly to a carbon atom of a benzene ring are called phenols. Alcohols and phenols dissociate in aqueous solutions as shown below.
ROH + H2O ⇌ RO– + H3O+
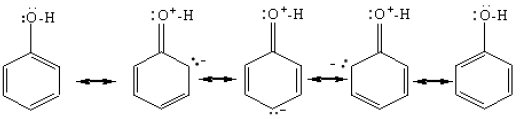
Phenols are more acidic than alcohols. This means that in the above equilibria, the equilibrium point for phenols is more towards the right than alcohols. The reason for this is that the stability of phenate ion relative to phenol is greater than the stability of the alkoxide ion relative to the alcohol. The phenate ion is more stable because its negative charge gets delocalized by resonance. In the alkoxide ion there is no such charge dispersion.
Reactions involving cleavage of the O-H bond
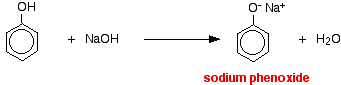
The higher acidity of phenols is confirmed by the following examples too. Although an alcohol reacts with sodium it does not react with NaOH. But phenol reacts with sodium as well as with NaOH. However, phenol is not acidic enough to react with Na2CO3.
2 C6H5OH + 2Na → 2C6H5O– Na++ H2
Non occurance of nucleophilic substitution reactions by breaking C-O bond
Unlike alcohols phenols do not undergo nucleophilic substitution reactions because,
(i) the C-O bond is shorter and stronger due to delocalization of lone pair of electrons
on the oxygen atom into the benzene ring. This can be shown by resonance.

(ii) phenyl cation is unstable.
The effect of the -OH group on the reactivity of the benzene ring in phenol.
• Due to the delocalization of the lone pairs of electrons which were on the oxygen atom with the benzene ring, the ring is rich with electrons. It has become very reactive towards electrophilic reagents. The O-H group of phenol is ortho, para directing. (It is not necessary to explain why the -OH group is ortho, para directing)
• When the electrophilic substitution reactions of phenol are compared with the corresponding reactions of benzene along with the relevant conditions, it is clear that the benzene ring of phenol, had become more reactive towards electrophiles.
Consider the following examples.
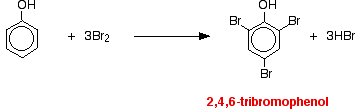
(i) Reacts immediately with bromine water to give a white precipitate of 2,4,6 – tribromophenol.
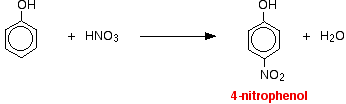
(ii) For nitration of phenol even dilute HNO3 is sufficiently reactive.
With dilute nitric acid
Phenol reacts with dilute nitric acid at room temperature to give a mixture of 2 nitrophenol and 4-nitrophenol.


With concentrated nitric acid
With concentrated nitric acid, more nitro groups substitute around the ring to give 2,4,6-trinitrophenol

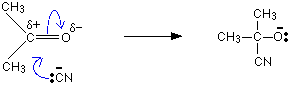
Mechanism of the addition of HCN to aldehydes and ketones.
This reaction is carried out by adding a dilute mineral acid into a mixture of the carbonyl compound and an aqueous solution of sodium cyanide. Here the CN ion acts as the nucleophile.
The mechanism for the addition of HCN to propanone
In the first stage, there is a nucleophilic attack by the cyanide ion on the slightly positive carbon atom.

The negative ion formed then picks up a hydrogen ion from somewhere – for example, from a hydrogen cyanide molecule.

The hydrogen ion could also come from the water or the H3O+ions present in the slightly acidic solution
The mechanism for the addition of HCN to ethanal
As before, the reaction starts with a nucleophilic attack by the cyanide ion on the slightly positive carbon atom.

It is completed by the addition of a hydrogen ion from, for example, a hydrogen cyanide molecule.

Mechanism of the reaction with Grignard reagent (RMgX)
![]()
![]()
R group of Grignard reagent together with the electron pair of R-Mg bond reacts as a nucleophile with carbonyl carbon.
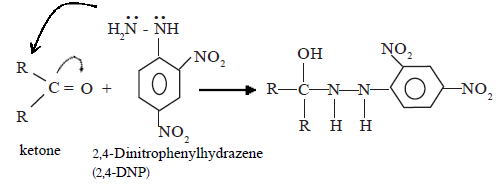
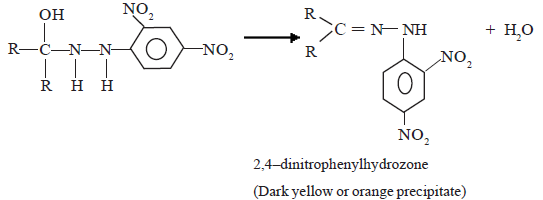
Mechanism of the reaction with Brady’s reagent (2,4-DNP)
(i) Nucleophilic addition
![]()
(ii) Dehydration
The above intermediate product undergoes dehydration as soon as it is formed to give the final product, which is 2,4–dinitrophenylhydrozone.
![]()
This reaction is used as a test to identify aldyhydes and ketones.
Reduction
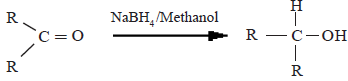
(i) Reduction by LiAlH4 or NaBH4
Here, the aldehydes and ketones get reduced to alcohols.
![]()
![]()
(ii) Reduction by Zn(Hg)/conc. HCl (Clemenson reduction)
Here, the aldehydes and ketones can be reduced to the corresponding hydrocarbons.
![]()
Oxidation of aldehydes
Aldehydes are oxidized to carboxylic acids even by mild oxidizing agents such as Tollen’s reagent and Fehling solution.
(i) Oxidation by Tollen’s reagent
Tollen’s reagent, [Ag(NH3)2]+ is prepared by adding dilute ammonium hydroxide to the precipitate of silver oxide formed by the addition of a few drops of dilute sodium hydroxide to an aqueous solution of silver nitrate.

Oxidation by Tollen’s reagent or the silver mirror test is used to distinguish between aldehydes and ketones.
(ii) Oxidation by Fehling solution
A solution of basic cupric tartarate is known as Fehling solution. This is a dark blue aquous solution. When a few drops of an aldehyde are added to this reagent and heated, the blue colour of the solution gradually disappears and a brick red precipitate of cuprous oxide is formed.

Aldehydes and ketones can be distinguished from each other by Fehling solution.
(iii)Oxidation by oxidizing agents such as acidified potassium dichromate or acidified chromic oxide or acidified potassium permanganate.
Aldehydes get oxidised to carboxylic acids by oxidizing agents such as acidified potassium dichromate or acidified chromic oxide or acidified potassium permanganate.

In the presence of aldehydes the pink colour of H+/KMnO4 solution become colourless. The orange colour of H+/Cr2O72- solution turns green. By using these reagents also aldehydes and ketones can be distinguished from each other.
The reactivity of the alpha position of aldehydes and ketones as exemplified by selfcondensation reactions.
• Due to the strong electron withdrawing nature of the carbonyl group, H atoms attached to the carbon atoms directly bound to the carbonyl carbon (the α- H) become acidic. The α- H can be abstracted as a proton by base (eg. OH–).The carbanion so formed is stabilized by resonance as shown below
![]()
The above carbanion attacks the carbon atom of the carbonyl group of an unionized aldehyde molecule as a nucleophile. Hence aldehydes and ketones with α hydrogens undergo self – condensation reactions.
Examples
Reaction of acetaldehyde in the presence of aqueous NaOH
![]()
Condensation of acetone
![]()
The addition products obtained above undergo dehydration easily.
Examples :
![]()
• The functional group of the carboxylic acids is the carboxyl group. In the carboxyl group there is a carbonyl group and a hydroxyl group attached to that carbon atom.
• Carboxyl group is a polar functional group. Due to the polarity of C – O and O – H groups it forms intermolecular hydrogen bonds. Relative to the aldehydes and ketones with equal relative molecular masses, the boiling points of carboxylic acids are higher.
• Carboxylic acids of C1 to C4 dissolve well in water. When the number of carbon atoms increases solubility decreases. Aromatic carboxylic acids are water insoluble and exist as solid crystalline substances. Almost all the carboxylic acids are soluble in organic solvents.
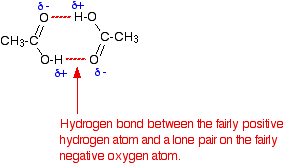
• The ability to form dimeric structures where carboxylic acid molecules are attached by hydrogen bonds as pairs is also a reason for the high boiling points of carboxylic acids.
Existence of carboxylic acids as dimer structures attached by hydrogen bonds is shown below.

Reactions of carboxylic acids
Reactions involving the cleavage of O – H bond




The above compounds react with Na2CO3 in the same way that they react with NaHCO3.
The acidic strengths of alcohols, phenols and carboxylic acids vary as follows.
Alcohols < Phenols< Carboxylic acids
The carboxylic acids attain the following equilibrium in an aqueous solution
![]()
The equilibrium point of the above equilibrium is situated more shifted towards the right side relative to the corresponding equilibrium attained by the phenols. The reason for this is that the stability of the carboxylate ion relative to the carboxylic acid is greater than the stability of the phenoxide ion relative to phenol. The carboxylate ion is a resonance hybrid of the following structures.
![]()
The stability of the carboxylate ion is due to the delocalization of the negative charge between two equivalant electronegative oxygen atoms.
Reactions involving cleavage of the C – O bond
• With PCl3/ PCl5
• With alcohols
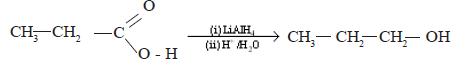
• Reduction of carboxylic acids with LiAlH4
Carboxylic acids give alcohols when reduced with LiAlH4 which is a very strong reducing agent. Carboxylic acids and their derivatives are not reduced by NaBH4.
![]()
Physical state and smell
Haloalkanes are colorless, sweet-smelling liquids. The lower members like methyl chloride, methyl bromide and ethyl chloride are colorless gases while members having very high molecular masses are solids.
Solubility
Haloalkanes are not able to form hydrogen bonds with water and, even though they are polar in nature, they are practically insoluble in water. However, they are soluble in organic solvents like alcohol, ether, benzene, etc.
Density
Chloroalkanes are lighter than water while bromides and alkyl iodides are heavier. With the increase in the size of the alkyl group, the densities go on decreasing in the order of :
fluoride > chloride > bromide > iodide.
Boiling points
The boiling points of alkyl chlorides, bromides and iodides follow the order RI > RBr > RCl where R is an alkyl group. With the increase in the size of halogen, the magnitude of Van der Waals forces increases and, consequently, the boiling points increase. Also, for the same halogen atom, the boiling points of haloalkanes increase with increase in the size of alkyl groups.
Alkyl halides are named as primary, secondary or tertiary depending on the number of H atoms attached to the carbon atom which carries the halogen atom.
primary
secondary
tertiary
Due to the high electronegativity of the halogen atom relative to the carbon atom, the C – X bond is polarized. As a result, there is a deficiency of electrons in that carbon atom. Therefore, nucleophiles attack this position. Nucleophiles are reagents which show a tendency to attack carbon nuclei and which are basic and rich in electrons. A nucleophile will bring a pair of electrons to form a bond with carbon.
Examples- OH– , CN– , R-C≡C– , R-O–
Nucleophilic substitution reactions are characteristic of alkyl halides. Here, the carbon atom forms a new bond with the nuclephile and the halogen atom leaves as a halide ion.
As a nucleophile possesses a pair of electrons available to form a new bond any nucleophile can also act as a base by forming a bond with H+ . Therefore, when an alkyl halide is reacted with a reagent such as :OH– , it can also undergo an elimination reaction by the mechanism shown below.
Here, :OH– instead of reacting as a nucleophile with carbon, reacts as a base and removes a H+ from the carbon atom adjacent to the carbon atom bearing halogen. The hydrogen atoms attached to the carbon atom adjacent to the carbon atom bearing the halogen atoms, have a low acidity due to the polarization of the C-X bond. Thus substitution and elimination are competing reactions in alkyl halides.
Alkyl halides react with Mg in the medium of dry ether to form the Grignard reagent
When an alkyl halide forms a Grignard reagent the polaritiy of the carbon atom originally joined to halogen, changes as shown below.
Due to the polarization of bonds in RMgX, the carbon attached to magnesium acts as a strong nucleophile and a very strong base.
RMgX + H2O → X-Mg-OH + RH
The strong basic character of the Grignard reagent can be shown by the following reactions.
• To explain the nucleophilic substitution reaction of alkyl halides, the time interval between bond breaking and bond making steps can be considered.
• When the breaking of the C-X bond and the formation of the new bond to the nucleophile takes place simultaneously, the nucleophilic substitution reaction of the alkyl halide takes place as a one step reaction.
Accordingly, the one step reaction can be presented as follows:
![]()
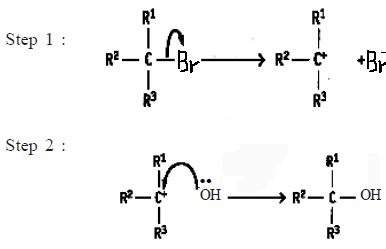
• When the formation of the new bond to the nucleophile takes place after the breaking of the C-X bond the nucleophlic substitution reaction of the alkyl halide takes place as a two step reaction.
Accordingly, the reaction that takes place by two steps can be presented as follows.
![]()
The reaction that takes place by two steps goes through an intermediate carbocation. On considering the stability of the carbocation formed, the tertiary alkyl halides (R1, R2, R3 = alkyl) which are able to form a more stable tertiary carbocations undergo nucleophilic substitution in two steps. The primary alkyl halides (R1, R2 = H, R3 = H or alkyl) undergo nucleophilic substitution reactions in one step as they are unable to form a stable intermediate carbocation.
The pathway taken by the secondary alkyl halides (R1 = H, R2, R3 = alkyl) depend on the reaction conditions.
Vinyl and phenyl carbocations are unstable and therefore, they do not react by the two step pathway. They also do not react by the one step path because the C-X bond is stronger than in alkyl halides due to its double bond character. This can be shown by resonance.
![]()
![]()
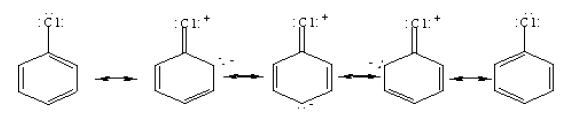
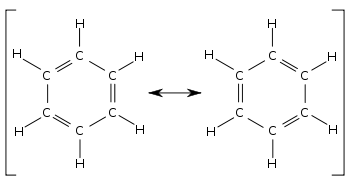
• Under normal conditions benzene does not answer the tests for unsaturation. Therefore, benzene cannot have a structure similar to that of a simple alkene or an alkyne.
• Although the structure proposed for benzene by Kekule shows three double and three single bonds for the molecule, the bond length between any two adjacent carbon atoms in benzene is the same.
• The carbon – carbon bond length of benzene is 1.39 x 10-10 m which is in between the length of carbon – carbon double bond (1.34 x 10-10 m) and the length of carbon – carbon single bond (1.54 x 10-10 m).
• Therefore, the structure of benzene is considered to be a hybrid of the resonance structures given below.

• The structure of benzene can be explained by molecular orbital theory. All its C atoms have undergone sp2 hybridization. Each carbon bears an unhybridized p orbital which can overlap with the unhybridized p orbitals on either sides of it.
• From this, a cyclic delocalized electron cloud is formed.
• Hence, the real structure of benzene is considered to be a hybrid of two Kekule structures.
• The real structure of benzene with delocalized electrons is more stable than the Kekule structure with three double bonds.
• The data for the standard enthalpy of hydrogenation helps to illustrate the stability of a benzene molecule.
The enthalpy change during this reaction is -120 kJ mol-1
The enthalpy change during this reaction is -208 kJ mol-1
But if benzene possesses three double bonds, its standard enthalpy of hydrogenation should be 3 x (-120 kJ mol-1
), that is -360 kJ mol-1
. Hence, benzene is more stable than its Kekule structures by an amount equal to (360-208) = 152 kJ mol-1
Electrophilic substitution reactions of benzene
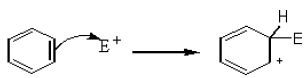
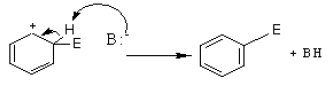
There is a loosely bound cloud of electrons on both faces of the planar benzene molecule. As with alkenes, this makes benzene reactive towards electrophiles. The first step in this reaction is for the electrophile (E+) to form a bond with a carbon atom in the benzene ring.
![]()
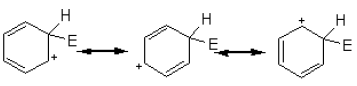
The intermediate carbocation so formed is stabilized by the delocalization of the positive charge by conjugation with the two π bonds. This can be shown by resonance.
![]()
However, in going from benzene to the carbocation the cyclic delocalization of π electrons is broken, and the aromatic stabilization energy is lost. Therefore, the intermediate carbocation prefers to lose a proton and re-established the cyclically delocalized electron cloud, rather than reacts with a nucleophile and give an addition product as in the case of alkenes.
The proton is usually taken up by one of the bases (B:–) present in the reaction mixture. Thus, the result is the substitution of a H atom on the benzene ring with E.
![]()
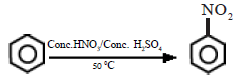
(i) Nitration
In the presence of the nitration mixture composed of conc. HNO3 and conc. H2SO4, nitrobenzene is formed by the substitution of H by a nitro group.
![]()
The electrophile here is +NO2 generated in the medium by the dehydration of nitric acid by sulphuric acid.
![]() The base which takes up the proton in the final step is HSO4–.
The base which takes up the proton in the final step is HSO4–.
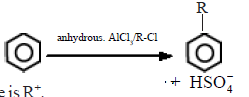
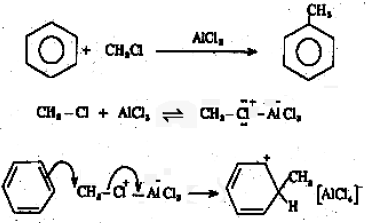
(ii) Friedel – Crafts alkylation
During the reaction of benzene with alkyl halides in the presence of a Lewis acid like anhydrous AlCl3, substitution by an alkyl group takes place.
![]()
The electrophile here is R+.
R-Cl + AlCl3 → R+AlCl4–
In cases where R+ is not very stable (eg. +CH3) the species actually reacting with the benzene molecule may be a R-Cl molecule polarized by coordination to AlCl3, which will transfer R+ to the benzene molecule during the reaction by cleavage of the R-Cl bond.
![]()
The base which takes up the proton in the final step is AlCl4–
AlCl4– + H+ → AlCl3 + HCl
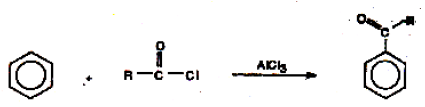
(iii) Friedel – Crafts acylation
During the reaction of benzene with acid chlorides in the presence of a Lewis acid like anhydrous AlCl3, substitution by an acyl group takes place
![]()
The electrophile here is R-C+=O.

The base which takes up the proton in the final step is
AlCl4– + H+ → AlCl3 + HCl
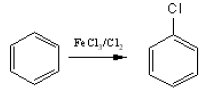
(iv) Halogenation
When benzene reacts with halogens (Cl2 or Br2) in the presence of a Lewis acids (such
as FeCl3, AlCl3) substitution by a halogen group takes place in the benzene ring.
![]()
The efective electrophile here is Cl+. It is transferred to the benzene ring from the complex AlCl4– during the reaction.
AlCl3 + Cl2 → Cl3Al––+Cl-Cl
![]()
The base which takes up the proton in the final step is AlCl4– .
AlCl4– + H+ → AlCl3 + HCl
Oxidation
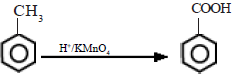
Benzene does not get oxidized by normal oxidizing agents like H+/KMnO4. However, the alkyl group in alkyl substituted benzene can be oxidized by H+/KMnO4 to a carboxylic acid group. The benzene ring does not oxidize easily due to its stability. H+/K2Cr2O7 can also be used for this reaction.
![]()
Tertiary alkyl groups do not get oxidized under the conditions in which primary and secondary alkyl groups get oxidized. More vigorous conditions under which tertiary alkyl groups can be oxidized also result in cleavage of the benzene ring.
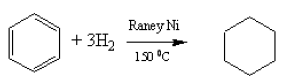
Catalytic hydrogenation
Although benzene does not undergo electrophilic addition reactions, like alkenes, they can add hydrogen in the presence of suitable catalysts. The temperatures used are higher than for alkenes.
![]()
Examples of activating groups in the relative order from the most activating group to the least activating:
-NH2, -NR2 > -OH, -OR> -NHCOR> -CH3 and other alkyl groups
with R as alkyl groups (CnH2n+1)
Examples of deactivating groups in the relative order from the most deactivating to the least deactivating:
-NO2, -CF3> -COR, -CN, -CO2R, -SO3H > Halogens
with R as alkyl groups (CnH2n+1)
The order of reactivity among Halogens from the more reactive (least deactivating substituent) to the least reactive (most deactivating substituent) halogen is:
F> Cl > Br > I
The order of reactivity of the benzene rings toward the electrophilic substitution when it is substituted with a halogen groups, follows the order of electronegativity. The ring that is substituted with the most electronegative halogen is the most reactive ring ( less deactivating substituent ) and the ring that is substituted with the least electronegatvie halogen is the least reactive ring ( more deactivating substituent ), when we compare rings with halogen substituents. Also the size of the halogen effects the reactivity of the benzene ring that the halogen is attached to. As the size of the halogen increase, the reactivity of the ring decreases.
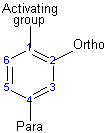
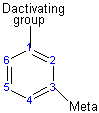
The direction of the reaction
The activating group directs the reaction to the ortho or para position, which means the electrophile substitute the hydrogen that is on carbon 2 or carbon 4. The deactivating group directs the reaction to the meta position, which means the electrophile substitute the hydrogen that is on carbon 3 with the exception of the halogens that is a deactivating group but directs the ortho or para substitution.

Substituents determine the reaction direction by resonance or inductive effect
Resonance effect is the conjugation between the ring and the substituent, which means the delocalizing of the ππ electrons between the ring and the substituent. Inductive effect is the withdraw of the sigma ( the single bond ) electrons away from the ring toward the substituent, due to the higher electronegativity of the substituent compared to the carbon of the ring.
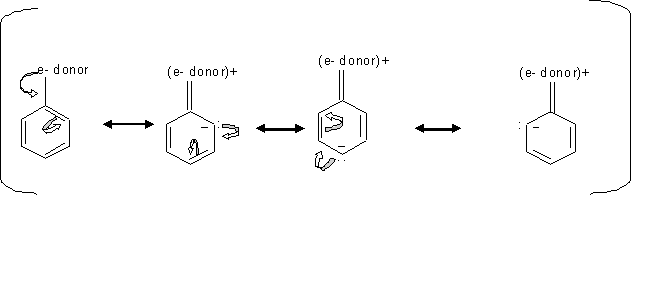
Activating groups (ortho or para directors)
When the substituents like -OH have an unshared pair of electrons, the resonance effect is stronger than the inductive effect which make these substituents stronger activators, since this resonance effect direct the electron toward the ring. In cases where the subtituents is esters or amides, they are less activating because they form resonance structure that pull the electron density away from the ring.

By looking at the mechanism above, we can see how groups donating electron direct the ortho, para electrophilic substition. Since the electrons locatinn transfer between the ortho and para carbons, then the electrophile prefer attacking the carbon that has the free electron.
Inductive effect of alkyl groups activates the direction of the ortho or para substitution, which is when s electrons gets pushed toward the ring.
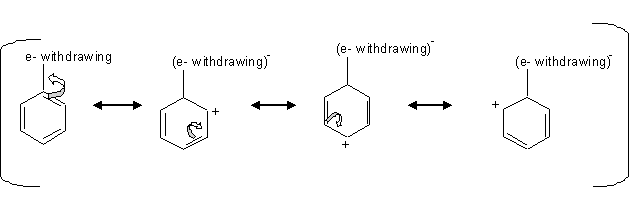
Deactivating group (meta directors)
The deactivating groups deactivate the ring by the inductive effect in the presence of an electronegative atom that withdraws the electrons away from the ring.

we can see from the mechanism above that when there is an electron withdraw from the ring, that leaves the carbons at the ortho, para positions with a positive charge which is unfavorable for the electrophile, so the electrophile attacks the carbon at the meta positions.
Halogens are an exception of the deactivating group that directs the ortho or para substitution. The halogens deactivate the ring by inductive effect not by the resonance even though they have an unpaired pair of electrons. The unpaired pair of electrons gets donated to the ring, but the inductive effect pulls away the s electrons from the ring by the electronegativity of the halogens.
Substituents determine the reactivity of rings
The reaction of a substituted ring with an activating group is faster than benzene. On the other hand, a substituted ring with a deactivated group is slower than benzene.
Activating groups speed up the reaction because of the resonance effect. The presence of the unpaired electrons that can be donated to the ring, stabilize the carbocation in the transition state. Thus; stabilizing the intermediate step, speeds up the reaction; and this is due to the decrease of the activating energy. On the other hand, the deactivating groups, withdraw the electrons away from the carbocation formed in the intermediate step, thus; the activation energy is increased which slows down the reaction.
The CH3Group is and ortho, para Director
Alkyl groups are Inductive activators
With o/p attack the form a tertiary arenium carbocation which speeds up the reaction


The O-CH3 Group is an ortho, para Director

Ortho and Para producst produces a resonance structure which stabilizes the arenium ion. This causes the ortho and para products for form faster than meta. Generally, the para product is preferred because of steric effects.
Acyl groups are meta Directors
Acyl groups are resonance deactivators. Ortho and para attack produces a resonance structure which places the arenium cation next to and additional cation. This destabilizes the arenium cation and slows down ortho and para reaction. By default the meta product forms faster because it lacks this destablizing resonance structure.

• Some features of carbon that contribute to the formation of a large number of organic compounds are given below.
• Between two C atoms strong single bonds, double bonds and triple bonds can be formed. It has been observed that compared to Si which is in the fourth group to which C belongs, the C – C, C = C, C≡C and C – H and other bonds that C forms possess higher bond energies
The relevant information is given in the following table.
Bond Bond energy
C – C 346
C = C 610
C ≡ C 835
C – H 413
Si – Si 226
Si – H 318
• Carbon can form chains containing thousands of atoms and also rings of all sizes.
• A carbon atom can form 4 covalent bonds. Hence, it is seen that a large number of other atoms/groups can get attached to a carbon atom in a chain or ring and that this property contributes to the existence of a large variety of compounds.
• Carbon forms strong covalent bonds not only with other carbon atoms and hydrogen atoms, but also with other non metal atoms such as O, S, P, N, and halogens.
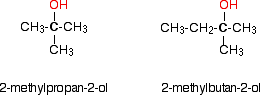
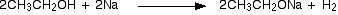
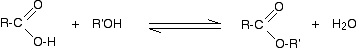
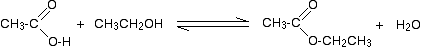

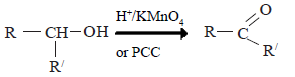

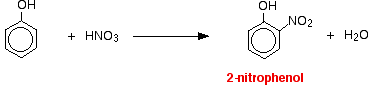
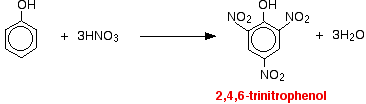
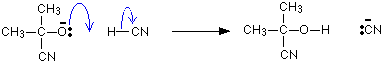
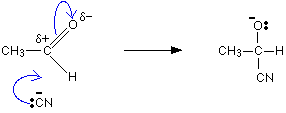
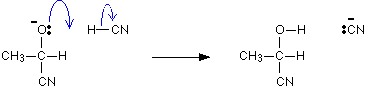
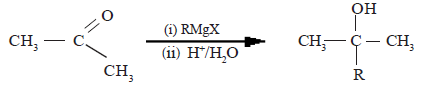
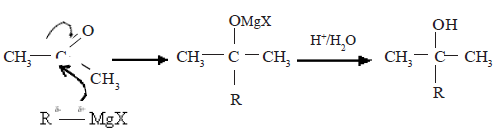
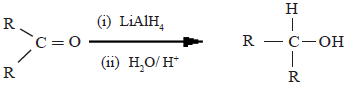
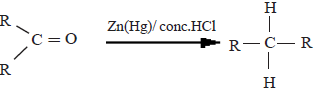
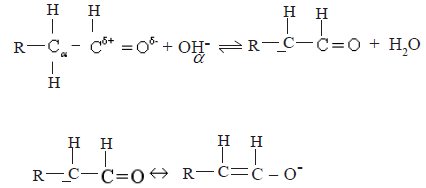


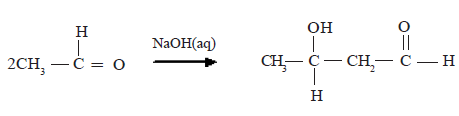
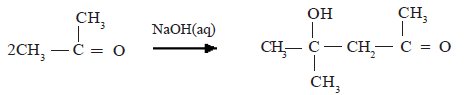
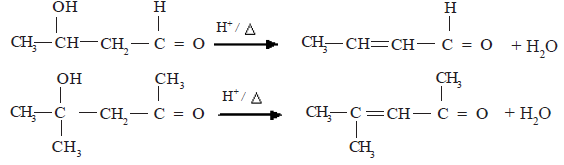
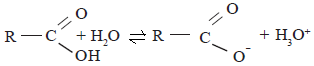
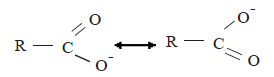
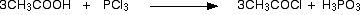
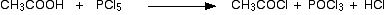
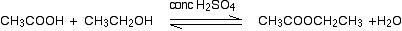
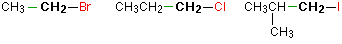
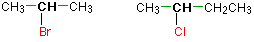
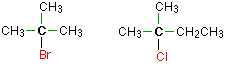
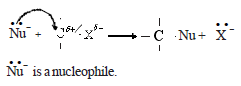
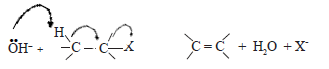
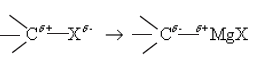
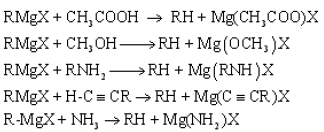
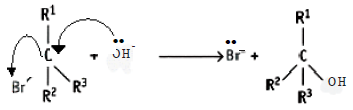
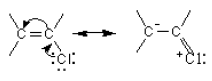
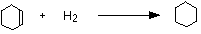 The enthalpy change during this reaction is -120 kJ mol-1
The enthalpy change during this reaction is -120 kJ mol-1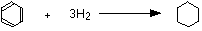 The enthalpy change during this reaction is -208 kJ mol-1
The enthalpy change during this reaction is -208 kJ mol-1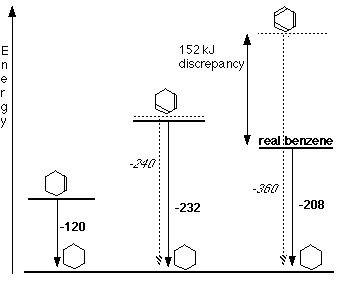
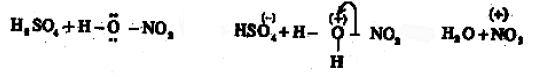
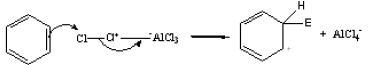












{kind=link}