• It is necessary to indicate concentration for solutions (e.g. Fe2+(aq, 1.0 mol dm--3) and pressure in case of gases e.g. H2(g, 1.0 atm).
When a piece of active metal such as magnesium is dipped in a beaker of water, there will be some tendency for the metal atoms to leave elctrons and go into solution as metal ions.The electrons will be left behind on the metal. In a very short time, there will be a build -up of electrons on the metal, and it will be surrounded in the solution by a layer of positive ions.These will tend to stay close because they are attracted to the negative charge on the piece of metal. This is called double layer as shown in figure (a).
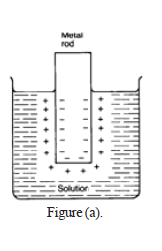
• Some of them will be attracted enough so that they will reclaim their electrons and stick back on to the piece of metal. A dynamic equilibrium will be established when the rate at which ions are leaving the surface is exactly equal to the rate at which they are joining it again. Here, the double layer reach to its equilibrium. At this point there will be a constant negative charge on the metal and a constant number of metal ions present in the solution around it. However, at the dynamic equilibrium, ions are continually leaving and rejoining the surface with equal rates. The potential that is created between the metal and adjecent layers of solution is known as the electrode potential.
• If a less active metal such as copper is considered, it forms its ions less readily. Any ions which do break away are more likely to reclaim their electrons and stick back on to the metal again. It will still reach an equilibrium position, but there will be a lesser charge on the metal, and fewer ions in the solution.
If both equilibria are considered together it is clear that the position of the magnesium equilibrium Mg2+(aq) + 2e ⇌ Mg(s) lies further to the left than that of the copper equilibrium Cu2+(aq) + 2e ⇌ Cu(s)
• By IUPAC convention, all these equilibria are written with the electrons on the left hand side of the equation. It is recommennded to stick on to this convention.
eg Calomel electrode
 Hg2Cl2(s) + 2e ⇌ 2Hg(s) + 2Cl–
Hg2Cl2(s) + 2e ⇌ 2Hg(s) + 2Cl–
eg Silver-Silver chloride electrode
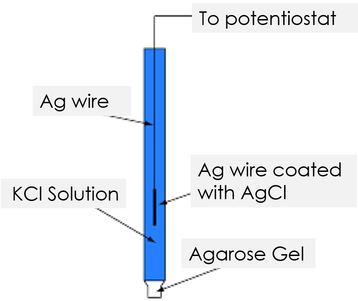 AgCl(s) + e ⇌ Ag(s) + Cl–
AgCl(s) + e ⇌ Ag(s) + Cl–
eg. Hydrogen electrode
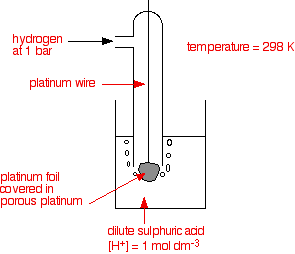 H+ + e ⇌ ½H2(g)
H+ + e ⇌ ½H2(g)
eg. Fe3+/Fe2+ electrode
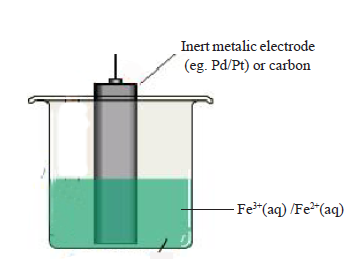
Fe3+ + e ⇌ Fe2+
• The anode, or oxidation half-cell is always written on the left; the cathode, or reduction halfcell,is written on the right. The two electrodes are electrically connected by means of a salt bridge, denoted by two vertical bars.
• The cell terminals are at the extreme ends in the cell notation. Electrode and electrolyte interface of each half cell is separated by a single vertical bar. Other constituents are separated by commas.
Examples: Pt(s)|Fe2+(aq, 1 mol dm-3) , Fe3+(aq, 1 mol dm-3)
Hg(l),Hg2Cl2(s)|Cl(aq, 1 mol dm-3)
• When the half-reaction involves a gas, an inert material such as platinum serves as a terminal and as an electrode surface on which half-reaction occurs. The notation for the hydrogen electrode, written as a cathode and as an anode are given below respectively.
H+(aq, 1 mol dm-3)|H2(g)|Pt(s) || Pt(s)|H2(g)|H+(aq, 1 mol dm-3)
• Cell notations of several cells are given below.
Zn(s)|Zn2+(aq,1 mol dm-3 )||Cu2+(aq, 1 mol dm-3)|Cu(s)
Cd(s)|Cd2+(aq, 1 mol dm-3)||H+(aq, 1 mol dm-3)|H2(g)|Pt(s)
• The cell in which the cell reaction cannot be reversed by providing electrical energy is called a “primary cell’.
eg-Daniel cell
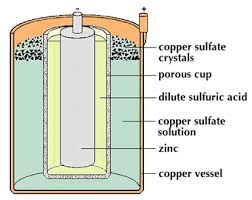
Electrolyte – NH4Cl/ZnCl2
(+) Pole – C/MnO2
(-) Pole – Zn
Reaction at (+) Pole(Cathode reaction) – 2NH4+(aq)+2MnO2(s)+2e → Mn2O3(s) + H2O(l)+2NH3(aq)
Reaction at (-) Pole(Anode reaction) – Zn(s) → Zn2+(aq) + 2e
Cell reaction – 2MnO2(s)+Zn(s)+H2O (l) → Zn2+(aq) + Mn2O3(s) +2OH–(aq)
Leclanche cell
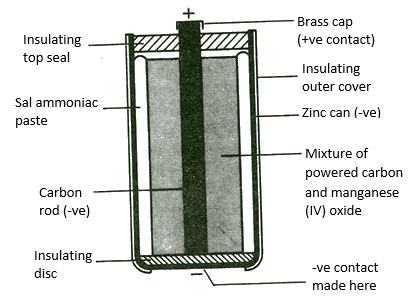
Electrolyte -ZnSO4/CuSO4
(+) Pole – Cu
(-) Pole – Zn
Reaction at (+) Pole(Cathode reaction) – Cu2+(aq) + 2e → Cu(s)
Reaction at (-) Pole(Anode reaction) – Zn(s) → Zn2+(aq) + 2e
Cell reaction – Zn(s) + Cu2+(aq) → Zn2+(aq) + Cu(s)
• A cell in which the reactants can be regenerated from the products by charging after
its discharge is referred to as a ‘secondary cell’.
eg. Lead accumulator

Electrolyte – dil. H2
SO4
Anode – Pb
Cathode – PbO2
Anode reaction during discharge – Pb(s) + SO42-(aq) → PbSO4(s) + 2e
Cathode reaction during discharge –
PbO2(s) + 4H+(aq) + SO42-(aq) + 2e → PbSO4(s) + 2H2O(l)
Electrochemcal cells in which reactants are continuously supplied from outside are referred as fuel cells. Dihydrogen and dioxygen, methane and dioxygen are commonly used for this purpose. One of the electrolyte used is concentrated KOH solution, maintained at 200 °C. Porous C-Ni electrodes are commonly used.
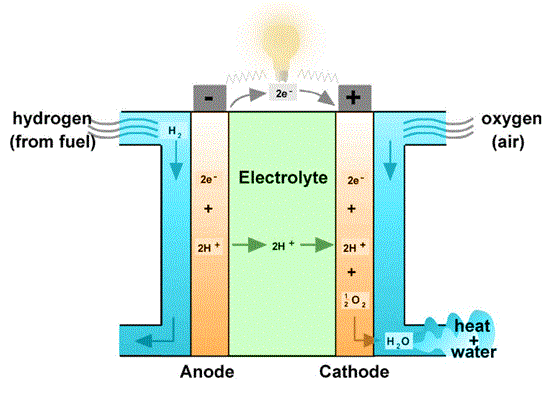
Relevent reactions for the dihydrogen-dioxygen fuel cell are given below.
Oxidation; 2H2(g) + 4OH–(aq) → 4H2O(l) + 4e
Reduction; O2(g) + 2H2O(aq) + 4e → 4OH–(aq)
Cell reaction 2H2(g) + O2(g) → 2H2O(l)
• Electrolysis is the passage of a direct current through a substance that is either molten or dissolved in a suitable solvent, resulting in chemical reactions at the electrodes and separation of mixtures.

• An ion-electron half reaction occurs at each electrode.
• The overall reaction is a redox reaction.
• The terminal at which oxidation occurs is the anode whereas the terminal at which reduction occurs is the cathode .
• The electrode connected to the positive terminal of the external source of electricity is the positive electrode. The electrode connected to the negative terminal is the negative electrode.
• The positive ions in the solution are attracted by the negative electrode. The negative ions are attracted by the positive electrode.
• Under certain conditions, a different species in the medium/set up may be oxidized or reduced in preference to the relevant ions.
Electrolysis of water (acidic or basic) using inert (carbon or platinum) electrodes
At anode; 2H2O(l) → O2(g) + 4H+(aq) + 4e
At cathode; 2H2O(l) + 2e → H2(g) + 2OH–(aq)
Electrolysis of aqueous CuSO4 solution using copper electrodes
At anode; Cu(s) → Cu2+(aq) + 2e
At cathode; Cu2+(aq) + 2e → Cu(s)
Electrolysis of aqueous CuSO4 solution using inert electrodes
At anode; 2H2O(l) → O2(g) + 4H+(aq) + 4e
At cathode; Cu2+(aq) + 2e → Cu(s)
Electrolysis of aqueous NaCl solution using inert electrodes
At anode; 2Cl–(aq) → Cl2(g) + 2e
At cathode; 2H2O(l) + 2e → H2(g) + 2OH–(aq)
Electrolysis of molten NaCl using inert electrodes
At anode; 2Cl–(l) → Cl2(g) + 2e
At cathode; Na+(l) + e → Na(l)
(1) The quantity of a substance liberated at an electrode is directly proportional to the quantity of electric charge that has flowed in the circuit.
(2) For a given quantity of electric charge, the amount of any metal deposited is proportional to its equivalent weight (atomic weight devided by the charge on the metal ion)
Faraday’s constant (F) = Molar charge of a proton = 1.602 x 10-19 C x 6.022 x 1023 mol-1= 96 484 C mol-1≅96 500 C mol-1
• Electroplating means coating a metal with another metal using electricity. This is different from the deposition of a less reactive metal on a more reactive metal.
In order to have a quality coating during electrolysis, the coating should strongly bind with the metal. In addition, it should have the following qualities.
• Strength
• Lustre
• Chemical inertness
• Good mechanical properties
• Absence of cracks and holes
• Uniformity in thickness and appearance
To get a quality metal coating the following factors should be appropriately controlled.
• The nature and purity of the electrolyte
• Concentration of ions
• Temperature
• Current density
• pH value
• Nature of the other ions presents
• Potential difference
• Purity of the anode
• Relative positioning of the anode and the cathode
• Cleanliness of the object and the nature of its surface
Two or more reactions may also occur simultaneously on the electrodes. They can be controlled by changing the temperature, concentration, voltage and the nature of electrodes.
In bimetallic corrosion, the more reactive metal corrodes faster in case where a pair of metals with electrical and physical contact is dipped in a corrosive electrolyte. The less corrosion resistant or the “active” member of the couple experiences accelerated corrosion while the more corrosion resistant or the “noble” member of the couple experiences reduced corrosion due to the “cathodic protection” effect. The most severe attack occurs at the joint between the two dissimilar metals. Further away from the bi metallic joint, the degree of accelerated attack is reduced.
It is a technique to control the corrosion of a metal surface by making it work as a cathode of an electrochemical cell. This is achieved by placing in contact with the metal to be protected another more easily corroded metal to act as the anode of the electrochemical cell.
Cathodic protection systems are most commonly used to protect steel, water or fuel pipelines and storage tanks, steel pier piles, ships, offshore oil platforms and onshore oil well casings.
Galvanizing of iron is an another example.
Protection of a metal by rendering its surface inactive by chemical methods is called passivation.
In the context of corrosion, passivation is the spontaneous formation of a hard non reactive surface film that inhibits further corrosion. This layer is usually an oxide or nitride that is a few molecules thick.
Stainless Steels can be passivated using a solution of nitric acid, to remove foreign particles from the surface and promote the growth of a protective oxide layer. Nickel can be used for handling elemental fluorine, due to a passivation layer of nickel fluoride.
• Strong electrolytes
Strong electrolytes are substances that are virtually fully ionized in solution, and include ionic solids and strong acids. As a result of their complete ionization, the concentration of ions in solution is proportional to the concentration of strong electrolyte added.
eg. NaCl, KNO3, HCl in aqueous solutions
• Weak electrolytes
Weak electrolytes are not fully ionized in solution. They include weak Bronsted acids and bases.
eg. CH3COOH, NH3, H2O
• Non-electrolytes
Liquids/solutions that do not contain ions are referred as non electrolytes. They do not conduct electricity.
eg. C6H6, Kerosene
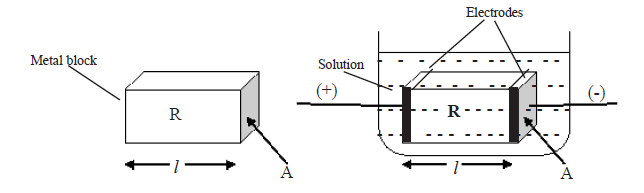
A metal cuboid is shown on the left hand side above. A cuboid shape portion of a solution with the similar dimensions of the metal cube which is in between two electrodes is shown on the right hand side.
Here l = Length of the metal cube or the selected cuboid shape portion of the solution (m)
A = Area of cross section (m2)
R = Resistance (Ω )
ρ= Resistivity (Ωm )
1/R = Conductance ( Ω-1 or S )
Regarding the above cuboids,
R∝l and R∝ (1/A)
∴ R∝(l/A)
R = ρl/A
ρ = RA/l
k =1/ρ = l/AR
• Coherent SI unit of conductivity is and the most practical unit is Ω-1m-1
• Conductivity and resistivity are constants for the particular substance (metal or solution with a given concentration) and it changes with temperature (about 2% per one degree Celsius in solution).
• Nature of the solute (aqueous solutions of strong, weak and non electrolytes, molten electrolytes)
• Concentration of the solute
• Temperature
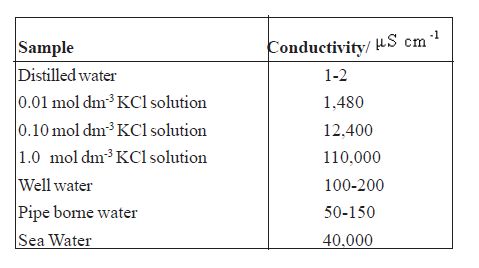
• For dilute solutions decrease in conductivity is approximately proportional to the concentration and this is very correct for very dilute solutions. The reason is the decrease of interactions among ions in dilution.
• Current = Charge / Time.
Current carried by an ion at a given temperature in a given electric field depends on the concentration of ions and their speed. Speed of an ion depends on its charge, size and potential gradient of the applied electric field.
• H+ and OH– ions have the highest speeds. So those ions contributes a lot for the conductance. For example, H+ ions contribute about 80% for the conductance of dilute HCl solution.
• The potential difference in the double layer of a given electrode is considered its electrode potential. This absolute difference cannot be measured. It can only be measured relative to another electrode.
• An electrode with a known or a defined potential used to measure the potential of a given electrode is called a reference electrode. Standard electrode potentials are measured with reference to the standard hydrogen electrode. The potential of the standard hydrogen electrode is defined as 0.00 V {[H+ (aq)] is 1.0 mol dm-3 and pressure of H2(g) is 1 atm and temperature is 298 K}. Calomel electrode and AgCl(s)|Ag(s) electrode are used as practical reference cells. Standard electrode potentials of Calomel electrode and AgCl(s)|Ag(s) electrode are +0.22 V and +0.2415V respectively.
• The value of the potential of a given electrode under standard conditions measured relative to the standard hydrogen electrode (SHE) is its standard electrode potential.
• The following diagram illustrates a set up that can be used for measuring the standard electrode potential of the Zn2+ (aq)|Zn(s) electrode.

• When the standard electrode potentials (reduction potential) of different electrodes are arranged in the ascending order, the electrochemical series is obtained.
Factors affecting the electrode potential
• Temperature
• Concentration of the electrolyte
• Nature of electrolyte
• Pressure (of a gas)
• Type of electrode
• Electromotive force (e.m.f.) is defined as the potential difference between the two electrodes when no current flows through the cell.
• Temperature, concentration of the electrolyte, electrode type and the nature of the electrolyte are the factors that affect the electromotive force. It is independent of the distance betweenthe electrodes and the surface area of the electrodes.
• Electrolytes in the two electrodes of an electrochemical cell are connected to complete the internal circuit through a salt bridge or a permeable membrane/diaphragm/porous partition.A tube filled with a solution of a salt such as KCl or NH4NO3 which is gelified with agar is used as a salt bridge. Such an electrochemical cell is known as a cell with a liquid junction.
• In a cell, when both electrodes are not separated by a salt bridge or a permeable membrane/diaphragm/porous partition is referred as a cell without a liquid junction.
• According to the international convention, electrode potentials are measured relative to the standard hydrogen electrode. On this standard, the electrode potential of the standard silver-silver chloride electrode is 0.22 V. Similarly, the electrode potentials of other standard electrodes can also be calculated. Show that in arranging these in the ascending order, electrochemical series is obtained.
Ecell = Ecathode – Eanode
When the rate of formation of a component in a system becomes equal to the rate of loss of it from the system, it is said to be in a steady state. This process can take place in open or closed systems. In a steady state, macroscopic properties do not change.
Examples :
(i) When the rate of inflow of water into a tank is equal to the rate of outflow, the volume of water in the tank remains the same.
(ii) When the ozone layer is considered, the concentration of ozone remains constant due to the following reactions, so it is at the steady state.
O3(g) →UV radiation O(g) + O2(g) → O3(g)
(iii) The concentration of O2 in the atmosphere remains constant due to various processes that release and consume oxygen.
(iv) Think of a candle burning uniformly. Apparently the flame does not change because the rates of entry and exit of materials to and from it are equal. The above systems are not in equilibrium.
Macroscopic properties
The properties experimentally determined or calculated taking a system as a whole are
macroscopic properties. Here no attention is paid to the particles constituting the system.
Consider the following reversible change taking place in a closed system with only A introduced into it at a constant temperature.
A ⇌ B
Initially, the rate of turning A into B is high and the rate of the reverse change is zero. With the formation of B, the rate of turning B into A increases and the rate of A becoming B decreases. At a certain moment, the rates of forward and backward reactions become equal. At this point, the system is in dynamic equilibrium. This can be shown by the graph given below.

• This kind of equilibria are established only if the change is reversible.
• The equilibrium is attained only in closed systems at constant temperature.
• The equilibrium can be approached starting from either end.
• The equilibrium is dynamic, i.e. both forward and reverse processes occur even at equilibrium at the same rate.
• The macroscopic properties of the system do not change at equilibrium.
• Dynamic equilibria are prevalent both in physical and chemical systems.
The following dynamic equilibrium is established between liquid water enclosed in a
closed container and water vapour in the space above it.
H2O(l) ⇌ H2O(g)
Iodine is a solid which sublimes. The following dynamic equilibrium sets in between iodine crystals and iodine vapour above them when some iodine is kept in a closed bottle.
I2(s) ⇌ I2(g)
The existence of an equilibrium between CO gas adsorbed on to charcoal and CO in the
gas phase inside a closed vessel is another example for this type of an equilibrium.
Consider a dilute solution formed by the dissolution of a gas like O2 in water. When this solution is in contact with a gaseous phase containing O2 such as the atmosphere, following equilibrium is established.
O2(solution) ⇌ O2(g)
These occur when a solute is distributed in two immiscible liquids in contact with each other. For instance, I2 dissolves both in water and CCl4. If some water is added to a solution of I2 in CCl4, iodine starts moving into the aqueous layer. With the increase of iodine concentration in the aqueous layer, iodine passes into the CCl4 layer and at a certain stage an equilibrium is established.
I2(H2O) ⇌ I2(CCl4)
These are equilibria encountered in relation to reversible chemical reactions.
N2(g) + 3H2(g) ⇌ 2NH3(g)
NH4Cl(s) ⇌ NH3(g) + HCl(g)
An ionic system should be a chemical system. Such systems contain ions. An equilibrium establishes between chemicals and ions in such systems.
H2O(l) ⇌ H+(aq) + OH–(aq)
AgCl(s) ⇌ Ag+(aq) + Cl–(aq)
CH3COOH(aq) ⇌ CH3COO–(aq) + H+(aq)
When a metal rod is immersed in a solution of its ions initially the metal atoms lose electrons and enter the solution as ions. The electrons reside in the metal. Consequently the metal becomes negatively charged. As the concentration of the metal ions in the solution increases, they capture electrons from the metal surface and become metal atoms. At a certain instant, the rate of ionization of metal atoms becomes equal to the rate of deposition of ions on the metal. At this equilibrium, the potential difference between the negatively charged metal surface and the positively charged metal ions in solution is called the electrode potential of the metal.
eg. Consider a zinc rod sunk in a Zn2+ ion solution.
Zn2+(aq) + 2e ⇌ Zn(s)
According to equilibrium law:
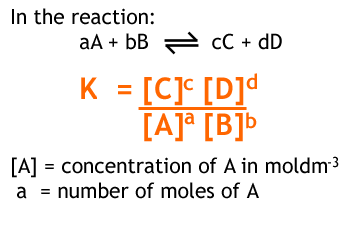
Where K is a constant called equilibrium constant. The symbol [ ] represents the concentrations of respective species at equilibrium.
• K depends only on temperature and is independent of initial and final concentrations of reactants and products. When expressing the equilibrium constant, the following requirements should be fulfilled.
• The relevant balanced equation for the equilibrium reaction should be given (Generally this is written so that the stoichiometric coefficient assume lowest whole numbers).
• The relevant temperature should be given.
• The physical states of the reactants and products should be given.
Attention should be paid to the following also.
• The equilibrium constant does not give any information with regard to the rate of the reaction.
• The value of K does not depend on initial concentrations.
• The value of K depends on temperature.
• By convention, the products are placed in the numerator of the expression.
• Since concentration of a solid or a pure substance is constant, they are incorporated into the equilibrium constant.
• The units of K depends on the expression for K (However, according to thermodynamic treatment, the equilibrium constant is dimensionless).
• The equilibrium constant expressed in terms of concentrations is known as Kc (‘c’ denotes concentration). The units of Kc depends on the stoichiometric equation for which the expression is written.
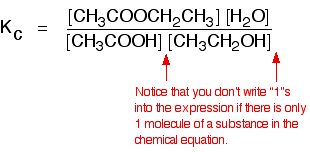
eg
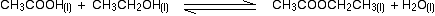
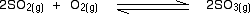
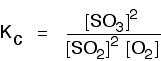
• If the gases in an equilibrium mixture behave ideally,
PV = nRT
n/V = P/RT
c = P/RT
∴At constant temperature, the concentration of a gas is proportional to its pressure. Partial pressure of a gas = mole fraction x total pressure
PG = PT x xG
Partial pressures are therefore proportional to the amount of gases and are used as a measure of concentration of a gas in a gaseous mixture.
• Consider the reaction between nitrogen gas and hydrogen gas, used to produce
ammonia as an example.
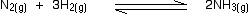
If the partial pressures of the individual gases in the equilibrium mixture are PH2,PN2 and PNH3 respectively then the equilibrium constant in terms of partial pressures Kp is given by expression,

• The partial pressures are treated as exact concentrations so they are raised to the power of the respective stoichiometric coefficient in the equation for the reaction.
• The value Kp describes the position of equilibrium and the concentrations of gases that can co-exist in an equilibrium.
• Reversible thermal dissociation in a closed system gives rise to an equilibrium mixture.
eg-In a closed system dissociation of dinitrogen tetraoxide into nitrogen dioxide
Heterogeneous equilibria
This equilibrium is only established if the calcium carbonate is heated in a closed system, preventing the carbon dioxide from escaping.
![]()
![]()
The only thing in this equilibrium which isn’t a solid is the carbon dioxide. That is all that is left in the equilibrium constant expression.
![]()
In a reaction,
aA + bB ⇌ cC + dD
we can write


Assuming the gaseous components to behave ideally,
PiVi = niRT
or

where (i) is the molar concentration of the species ‘i’.


n = (Number of moles of gaseous products)-(Number of moles of gaseous reactants).
Thus, n is equal to the difference in the number of gaseous moles of products and the number of gaseous moles of reactants. The above equation can be rewritten as
Kp = Kc (RT)Δn

• In a closed vessel at constant temperature, the following equilibrium exits between water and water vapour.
H2O(l) ⇌ H2O(g)
• Applying equilibrium law,
Since [H2O(l)] is a constant, K’ =[H2O(g)]
Since the concentration of a gas is proportional to its pressure at constant temperature, K = P0H2O
Where P0 is the saturated vapour pressure of water at the temperature concerned. Hence, the equilibrium constant of this system can be taken as the saturated vapour pressure of water at the temperature concerned
• It is the equilibrium constant describing the distribution of a solute species between two immiscible solvents.
• Immiscible liquids, if shaken together, mix temporarily but eventually separate into distinct phases with the most dense at the bottom and the least dense at the top. A visible boundary, the meniscus, separates two phases.
• The ratio of the concentration of a solute species that is distributed between two immiscible solvents at a given temperature is a constant.
If you have two immiscible liquids like ether and water, and shake them up in a separating funnel, they obviously form two layers. The ether is less dense than water, and so forms the top layer.
Now suppose you shake up a mixture of ether and water containing a substance which is soluble in both of them. Let’s suppose that the substance, X, is more soluble in ether than it is in water.
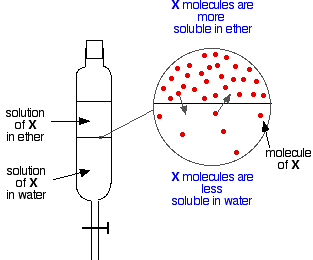
Particles of X will cross the boundary between the two liquid layers, and you will soon get a dynamic equilibrium set up. For every particle which moves into the top layer, one will move back down into the bottom one.
You could write an equation for this:
![]()
![]()
. . . and like any other equilibrium, you can find an equilibrium constant:
![]()
![]()
This equilibrium constant is called the partition coefficient, and is often given the symbol KD.
Like other equilibrium constants, partition coefficients are constant at a constant temperature, but they have some other restrictions as well. They only work properly for fairly dilute solutions, and the solute must be in the same chemical form in both solvents. It mustn’t react, or ionise or associate (join together in dimers, for example)
• The extent of a chemical reaction when equlibrium is established is called the position of equlibrium. This varies from one reaction to another and depends upon the temperatue at which the reaction is performed. The equlibrium constant is a measure of the position of an equlibrium. If equlibrium constant is more than one, then the position of equlibrium is said to lie towards the right.
• The position of equilibrium is not fixed for a reaction but changes as you change the reaction conditions. When a system which is in dynamic equilibrium is disturbed, it tends to respond in such a way as to minimize the disturbance and to restore equilibrium. This is known as Le Chatelier’s principle.
• The following factors change the equilibrium point.
– Concentration
– Pressure
– Temperature
Le Chatelier’s principle can be used to deduce the effect of change of those factors on equilibrium.
• Le Chatelier’s principle is a qualitative treatment of the equilibrium law. Whenever possible attempt must be made to explain facts quantitatively using equilibrium law.
Attention should be paid to the following.
1. Change in concentration of each substance.
2. Increasing the pressure of the system (Decrease in volume here can be considered as an increase in concentration).
3. Introduction of an inert gas or gas that does not affect the reaction, into the system.
4. The influence of temperature on the equilibrium constant is not discussed. Therefore, use Le Chaterlier’s principle to predict the results of changes in temperature takingexothermic/endothermic nature of the reaction.
5. A catalyst does not change the position of the equilibrium.
• Consider the molecules of a liquid A and its vapour are in random motion. If the liquid is in an open container, the molecules in the vapour phase spread. Consequently, more and more molecules enter the vapour phase from the liquid phase till the whole liquid evaporates.
• When the evaporation occurs in a closed space, molecules enter the vapour phase from the liquid phase and vice versa and a dynamic equilibrium is established between the two phases at the temperature concerned. At this equilibrium, the rate of evaporation is equal to the rate of condensation.
A(l) ⇌ A(g)
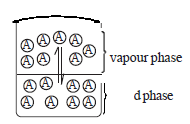
However, the microscopic changes are not observable. If the temperature remains unchanged the pressure exerted by the vapour at equilibrium remains constant and it is the saturated vapour pressure at the particular temperature. This itself is an equilibrium constant.
• The kinetic energy of molecules depends on the temperature. When the temperature rises, the kinetic energy increases and the molecules with high kinetic energy overcome the intermolecular forces and move into the vapour phase. The higher the intermolecular attractions, the lesser the tendency of the molecules to pass into the vapour phase.
• As more and more molecules move into the vapour phase, the vapour pressure increases. Hence with increasing temperature, the vapour pressure also increases. When the temperature is decreased, the kinetic energy of the molecules in the vapour phase decreases with the result that more molecules return to the liquid phase and the vapour pressure drops.
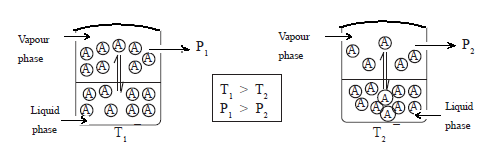
• When the liquid is heated, the temperature rises and at a certain temperature the saturated vapour pressure becomes equal to the (external) atmospheric pressure. At this temperature, the liquid boils and therefore, it is the normal boiling point of the liquid.
• The increase in vapour pressure of liquids with temperature is not linear . Moreover, different liquids have different intermolecular attractive forces and hence different volatilities. Therefore, the temperatures at which their s.v.p. becomes equal to the atmospheric pressure are different, so they boil at different temperatures
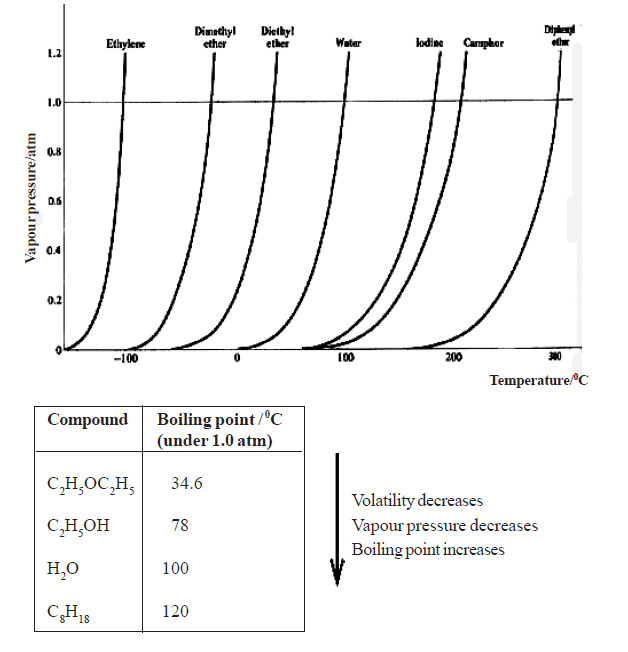
• Consider the liquid-vapour equilibrium established in a closed system. With rise in temperature the equilibrium is disturbed and more molecules pass into the vapour phase from the liquid phase establishing new equilibria increasing the amount in the gas phase. Finally a stage is reached where only the vapour exists.
• This vapour can be liquefied by compression. However, the tendency of the vapour to liquefy decreases with increasing temperature. Hence, a gas has a characteristic minimum temperature, called the critical temperature, above which the gas cannot be liquefied by increasing pressure, no matter how high the pressure would be.
The minimum pressure required to bring about liquefaction at the critical temperature is known as the critical pressure and the volume of one mole of the substance at the critical temperature and critical pressure is known as the critical volume.
• The following phase diagram of water shows the variation of vapour pressure of water with temperature (curve TC) and vapour pressure of ice with temperature (curve AT). Line BT shows the temperatures and pressures at which ice and liquid water are in equilibrium.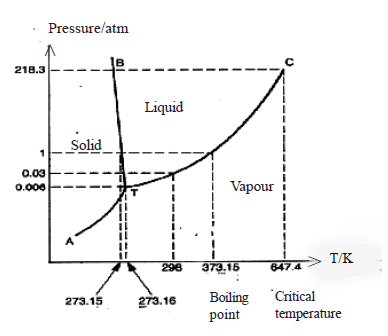
At the point T ice, water and vapour exist together in equilibrium, and this point is known as the triple point. The curve TC ends at the critical temperature (647.4 K) of water. Above this temperature only the vapour phase can exist.
• Liquid-liquid systems can be classified as
– Totally miscible liquid-liquid systems
e.g. water and ethanol, hexane and heptane, benzene and toluene.
– Partially miscible liquid-liquid systems
e.g. water and butanol, water and ether
– Totally immiscible liquid-liquid systems
e.g. water and tetrachloromethane
In the case of a binary solution composed of totally miscible liquid components A and B, it may be possible that f(A-B) = f(A-A) = f(B-B) where f denotes inter-molecular attractive forces or interactions. Such a solution is called an ideal solution. When the components of an ideal solution are mixed (a) the volume does not change and (b) the enthalpy does not change (and hence there is no observable change in temperature).
• When an ideal solution of A and B evaporates into a vacuum in a closed space, the molecules of both A and B with sufficient kinetic energy to surmount the liquid phase interactions escape from the surface into space above. At the same time, the molecules of A and B in motion in the vapour phase return to the liquid phase. When the rates of these two processes are equal, a dynamic equilibrium sets in. This is testified by the consistency of (a) the total vapour pressure (to which partial pressures of A and B contribute) and (b) the composition of the vapour phase at a constant temperature.
• The composition of the vapour depends on (a) the relative volatilities of A and B (and hence their boiling points) and (b) the relative concentrations of A and B in the solution.
• The higher the volatility and the higher the concentration of a certain component the greater is its tendency to be in the vapour phase and to exert a higher partial pressure.
• In order to find the composition of the vapour of an ideal binary solution quantitatively, let us consider the equilibria existing in a liquid-vapour system.
A(l) ⇌ A(g) ——-(1)
B(l) ⇌ B(g) ——-(2)
If R1 is the rate of moving A from liquid phase to gas phase
R1= k/ [A(l)]
Since [A(l)] is proportional to its mole fraction xA in the liquid phase,
R1= k1 × xA ——-(3)
If R2 is the rate of moving A from gas phase to liquid phase
R2= k” [A(g)]
Since [A(g)], is proportional to its partial pressure PA
R2= k2 × PA ———(4)
At equilibrium R1= R2
According to (3) and (4)
k2PA = k1xA
∴PA = (k1/k2)xA
∴PA = k xA
When xA = 1, PA = P0A
k = P0A
PA= P0A. xA
∴Similarly PB= P0B
Thus, in an ideal solution, the partial pressure of a given component A is equal to the product of the vapour pressure of pure A and the mole fraction of A in the liquid phase at constant temperature. This relationship is called Raoult’s law.
• It is understood that PA < P0A
and PB < P0B
Hence lowering in the vapour pressure of A = P0A– PA = P0A – P0A. xA =P0A (1 – xA ) =P0A. xB
∴ ( P0A– PA )/ P0A = xB
This is an alternate form of Raoults’ law.
• Combining Raoults’ law with Dalton’s law of partial pressures, makes it possible to determine the composition of the vapour phase. If PAB is the total vapour pressure and yA and yB are the mole fractions of A and B in the vapour phase respectively :
PA = PA yA
∴PA = (PA + PB ) yA
∴P0A. xA = (P0A. xA + P0B. xB) yA
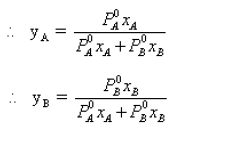
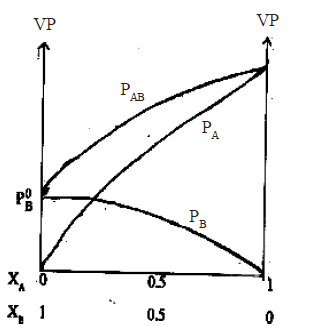
• According to Raoult’s law PA= P0A. xA . Since P0A is a constant at constant temperature,the graph of the partial vapour pressure of a component against its
mole fraction in an ideal solution is a straight line.
• Assuming A is more volatile than B(i.e. P0A > P0B and ∴ Tb(A) < Tb(B) the plot of vapour pressures versus mole fractions of an ideal solution will be as follows.
Here PAB is total pressure and PA and PB are vapour pressure of A and B respectively
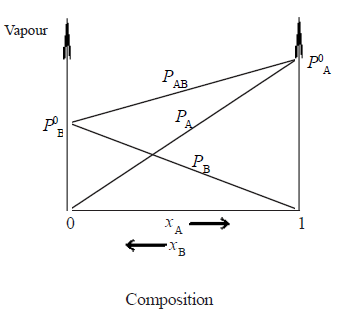
examples -Hexane and heptane, benzene and toluene, bromoethane and iodoethane, CCl4 and CHCl3,C6H6 and C6D6 (Note : PAB at a given composition is equal to PA + PB).
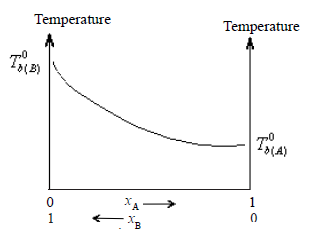
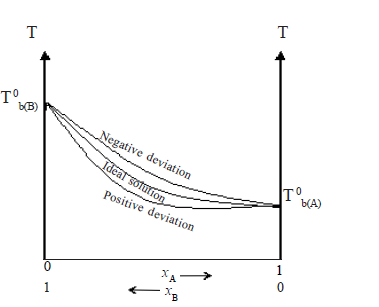
The boiling boint-composition graph, however, is not a straight line.
• Ideal solutions obey Raoults law. There are solutions which deviate from Raoult’s law. In these f(A-B) ≠ f(A-A) ≠ f(B-B). These are non ideal solutions.
• In some f(A-B) >f(A-A) , f(A-B) >f(B-B) and and the freedom for molecules to escape into the vapour phase is lesser than in the case of an ideal solution.
PA< P0A. xA
PB< P0B. xB
PAB < ( P0A. xA + P0B. xB )
Therefore, the curves in the vapour pressure – composition diagram dips downwards whereas the boiling point – composition curve rises up.
e.g. Propanone and methanol, trichloromethane and propanone, ethanoic acid and water.
These solutions are said to exhibit a negative deviation. When components of such solutions are mixed, temperature increases and volume decreases.
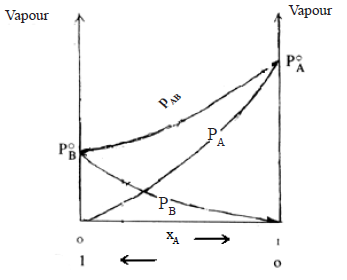
In some others f(A-B) < f(A-A) , f(A-B) < f(B-B) and the molecules tend to escape more freely into the vapour phase than in the case of an ideal solution
PA> P0A. xA
PB> P0B. xB
PAB > ( P0A. xA + P0B. xB )
Thus the curves in the vapour pressure – composition diagram loops upwards. The boiling point curve dips downwards.
e.g. Propanone and carbon disulphide, ethanol and benzene.
These solutions show positive deviations from Raoult’s law.
When solvents of such solutions are mixed, temperature decreases and volume increases.
• Simple distillation can be used to get pure water from a solution of salt in water.
• In addition to a distillation flask, for distillation a condenser is also need
• In simple distillation only one component enters the vapour phase.
Using the phase diagram
. 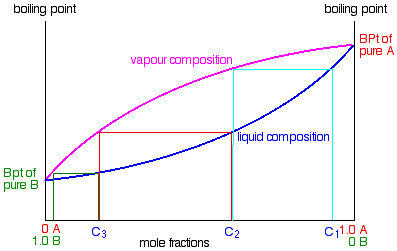
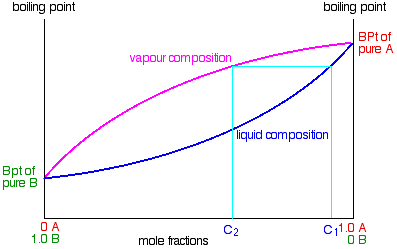
If you boil a liquid mixture C1, you will get a vapour with composition C2, which you can condense to give a liquid of that same composition (the pale blue lines).
If you reboil that liquid C2, it will give a vapour with composition C3. Again you can condense that to give a liquid of the same new composition (the red lines).
Reboiling the liquid C3 will give a vapour still richer in the more volatile component B (the green lines). You can see that if you were to do this once or twice more, you would be able to collect a liquid which was virtually pure B.
The secret of getting the more volatile component from a mixture of liquids is obviously to do a succession of boiling-condensing-reboiling operations.
The apparatus
A typical lab fractional distillation would look like this:
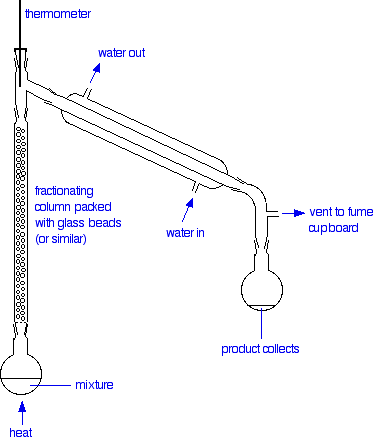
The fractionating column is packed with glass beads (or something similar) to give the maximum possible surface area for vapour to condense on. Some fractionating columns have spikes of glass sticking out from the sides which serve the same purpose.
The mixture is heated at such a rate that the thermometer is at the temperature of the boiling point of the more volatile component. Notice that the thermometer bulb is placed exactly at the outlet from the fractionating column.
Relating what happens in the fractionating column to the phase diagram
Suppose you boil a mixture with composition C1.
The vapour over the top of the boiling liquid will be richer in the more volatile component, and will have the composition C2.
That vapour now starts to travel up the fractionating column. Eventually it will reach a height in the column where the temperature is low enough that it will condense to give a liquid. The composition of that liquid will, of course, still be C2.
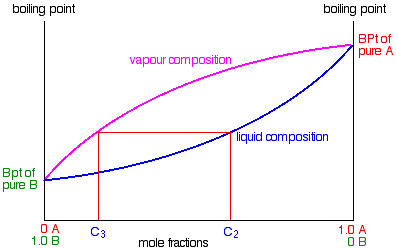
Some of the liquid of composition C2 will boil to give a vapour of composition C3. Let’s concentrate first on that new vapour and think about the unvaporised part of the liquid afterwards.
The vapour
This new vapour will again move further up the fractionating column until it gets to a temperature where it can condense. Then the whole process repeats itself.
Each time the vapour condenses to a liquid, this liquid will start to trickle back down the column where it will be reboiled by up-coming hot vapour. Each time this happens the new vapour will be richer in the more volatile component.
The aim is to balance the temperature of the column so that by the time vapour reaches the top after huge numbers of condensing and reboiling operations, it consists only of the more volatile component – in this case, B.
Whether or not this is possible depends on the difference between the boiling points of the two liquids. The closer they are together, the longer the column has to be.
The liquid
So what about the liquid left behind at each reboiling? Obviously, if the vapour is richer in the more volatile component, the liquid left behind must be getting richer in the other one.
As the condensed liquid trickles down the column constantly being reboiled by up-coming vapour, each reboiling makes it richer and richer in the less volatile component – in this case, A. By the time the liquid drips back into the flask, it will be very rich in A indeed.
So, over time, as B passes out of the top of the column into the condenser, the liquid in the flask will become richer in A. If you are very, very careful over temperature control, eventually you will have separated the mixture into B in the collecting flask and A in the original flask.
• The solubility of NaCl in water is approximately 5 mol dm-3. In the solution there are interactions between Na+ and Cl– ions. Though simple, they are not independent of one another.
• In the case of a sparingly soluble electrolyte such as BaSO4, the ions exist independently in the solution and the following dynamic equilibrium exists between the solid and the ions in a saturated solution.
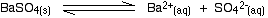
Applying the law of equilibrium
[BaSO4(s)] is a constant. Therefore,
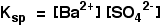
• Ksp is a constant at constant temperature and is called ‘solubility product’. If the solubility of BaSO4 is x mol dm-3, Ksp = x2 mol2 dm-6.
• In general, solubility product is not applied for salts having high solubility.
• If the ionic product is greater than the solubility product of a salt, it will precipitate.
eg. If [Ba2+][SO42-] > Ksp BaSO4 will precipitate.
Example
The solubility of magnesium hydroxide at 298 K is 1.71 x 10-4mol dm-3. Calculate the solubility product.
The equilibrium is:
![]()
![]()
For every mole of magnesium hydroxide that dissolves, you will get one mole of magnesium ions, but twice that number of hydroxide ions.
So the concentration of the dissolved magnesium ions is the same as the dissolved magnesium hydroxide:
![]() [Mg2+] = 1.71 x 10-4 mol dm-3
[Mg2+] = 1.71 x 10-4 mol dm-3
The concentration of dissolved hydroxide ions is twice that:
![]() [OH–] = 2 x 1.71 x 10-4 = 3.42 x 10-4 mol dm-3
[OH–] = 2 x 1.71 x 10-4 = 3.42 x 10-4 mol dm-3
Now put these numbers into the solubility product expression and do the sum.
![]()
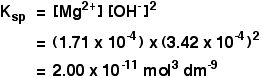
• On addition of NaCl solution to a saturated solution of AgCl, concentration of Cl– ions increases. Since Ksp is a constant at constant temperature Ag+ concentration in the medium should decrease. Therefore, AgCl is precipitated. In other words, the solubility of AgCl in a solution ofup> ions is less compared to its solubility in pure water. This is called common ion effect. This principle is applicable for the Ag+ ion as well.